Within the scope of the European Union Medical Device Regulation (MDR) published in May 2017; Clinical Studies for medical devices are specified as part of the “Clinical Evaluation Process”.
We believe that it would be beneficial to make preparations now for the "clinical studies" that we will hear about frequently in the coming years. For this reason, we think that it is important to get closer to the concepts and establish the general framework of the process to begin with, in terms of business continuity and compliance with the law.
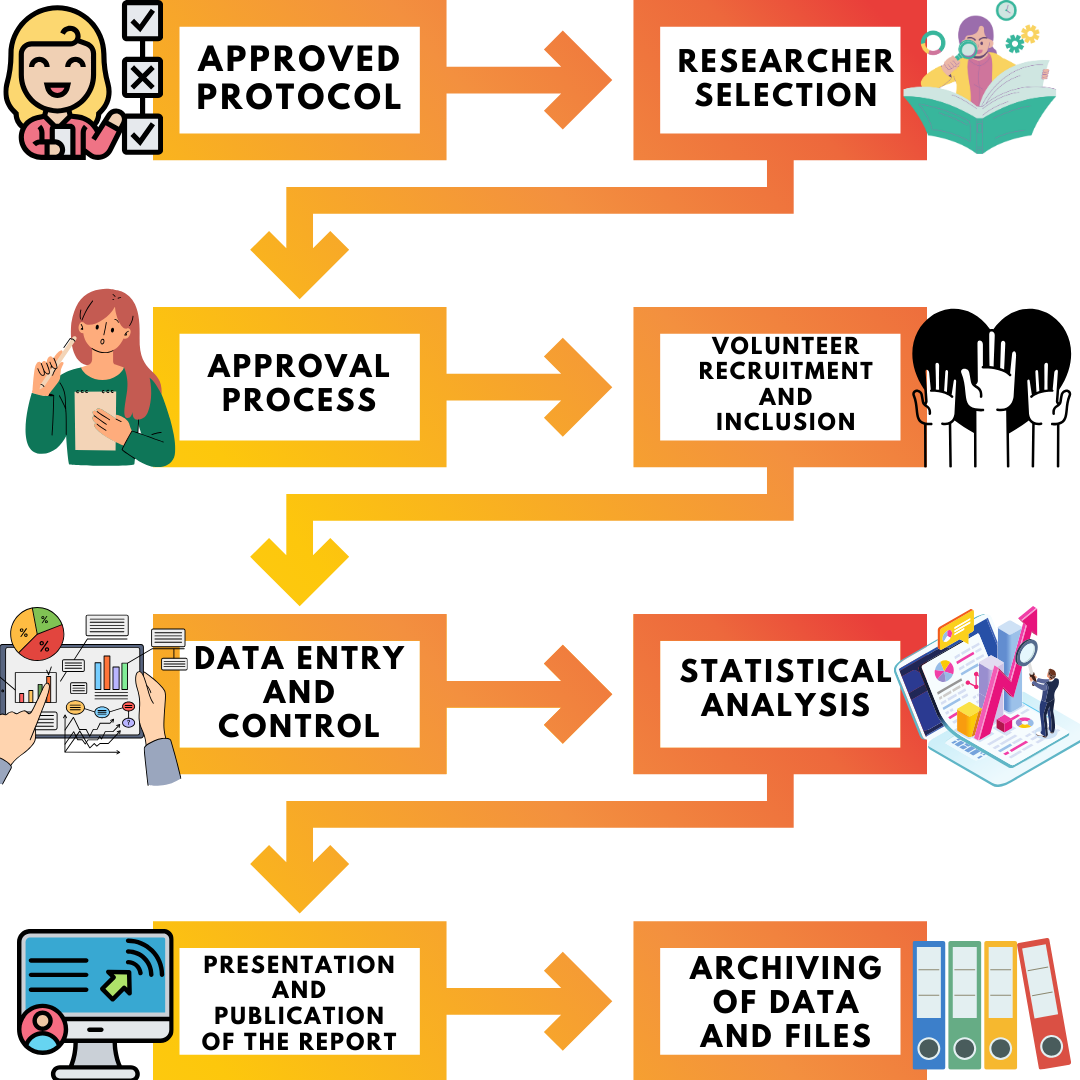
- In accordance with the MDR, approval from the Competent Authorities is required for the initiation of the clinical study, which must be carried out within the scope of the authorization of the Member State(s)
- The Ethics Committee established in accordance with the national law of the Member State where the clinical study will be conducted must not have expressed any negative opinion regarding the clinical study
- Within the European Union, there must be a established Sponsor or the Sponsor's legal representative or, depending on the regulation of the country in which the study will be conducted, a contact person
- Appropriate protection of the vulnerable population/volunteers (such as children, elderly, disabled) participating in the study must be ensured
- For volunteers or public health; The expected benefits, foreseeable risks or non-conformities need to be justified (a risk/benefit assessment in favor of the benefit and a justifiable research purpose justify the conduct of the study) and this compliance must be monitored on an ongoing basis
- In cases where the volunteer is underage or unable to give informed consent, the legal representative of the volunteer must give informed consent and provide contact information of an institution or legal entity from which the necessary information can be obtained in case of need
- Within the scope of the 95/46/EC Data Protection Directive, patients; their rights to physical and mental integrity and privacy must be protected
- The clinical study should be designed to have low levels of pain, suffering and predictable risk, as specifically stated in the Clinical Study Plan, and the risk threshold and degree of distress that the volunteers will be exposed to due to the study should be clearly defined within the scope of the plan
- Medical care within the clinical study must be provided by a suitably qualified medical doctor, or, where appropriate, a dentist or other person who can undertake medical care in clinical studies as specified under national law
- The main rule is not to make an improper offer to the subject or the subject's legal representative to participate in the clinical trial, including financial benefit
- It is requested that the device to be used in the study comply with general safety and performance principles in a way that does not endanger patient health, and that all precautions are taken to prevent possible dangers, and that the necessary technical, biological, safety and pre-clinical tests are proven by taking into account the state of the art
- The general conditions and documentation requirements included in MDR Annex 15 (application form, investigator brochure, available clinical data, clinical study plan) must be met.
> The documentation in Annex 15 shall be retained for at least 10 years from the end of the clinical study or, in cases where the device is placed on the market immediately thereafter, from the date of the last device being placed on the market. In the case of implantable devices, this period is at least 15 years.
> The sponsor has an existing agreement that adverse events related to the clinical study will be reported by the investigator or investigators in a timely manner.
> In order to monitor that the study is carried out in accordance with the Clinical Study Plan, Good Clinical Practices and MDR, the Sponsor; appoints an observer independent from the study centre.
> Since it is the Sponsor's responsibility to follow up the participants in the clinical study, it invites the volunteers to visit them at specified periods and completes their checks.
> The sponsor provides evidence in audits that the study was conducted in accordance with Good Clinical Practice.

- It should be possible for the subject or his/her legal representative to withdraw from the study by withdrawing informed consent at any time without prejudice and without providing any justification.
- The investigator must have the training and sufficient scientific competence and experience to provide the necessary medical care for patients within the scope of the clinical study. In addition, the researcher must be working in a professional field recognized by the Member State.
- Other personnel who will participate in the study must have appropriate competence in the relevant medical field and clinical study methodology in terms of experience or education.
- The centers where the study will be conducted must be suitable for clinical studies and have similar characteristics to the facilities where the device will be used.
Compliance with the above-mentioned conditions is evaluated by Member States. Member States may also require sterilization validation where the use of sterile devices is required or evidence of the sterilization procedures to be used at the study centre. Furthermore, where appropriate, evidence must be provided regarding the quality, safety and performance of substances of animal or human origin that can be assessed under 2001/83/EC.
We would like to remind you that the necessary legal regulatory preparations are continuing in our country and Member countries to conduct Clinical Studies in compliance with MDR.
In this process, we recommend that our manufacturers first examine in detail the clinical studies conducted and published on the products they have determined to be equivalent in their Clinical Data Evaluation reports.