Biological evaluation of medical devices
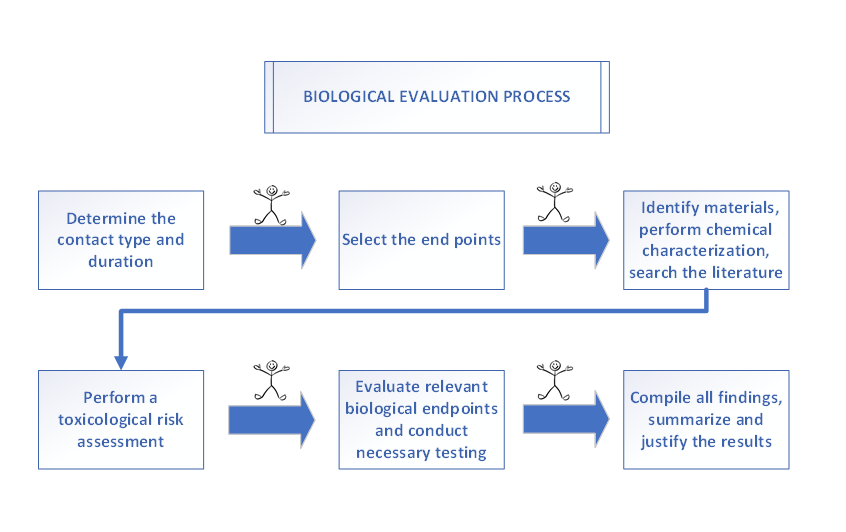
Before placing a medical device on the market, most of the regulative authorities require the manufacturers to thoroughly assess the device's biological safety. This ensures that any risk of device’s incompatibility with the human body is minimized. The ISO 10993 series outlines the requirements to effectively reduce biological risks to an acceptable benefit/risk ratio to demonstrate biocompatibility of medical devices. This biological evaluation process is closely intertwined with the risk management process and may involve pre-clinical testing through material characterization or specific biological testing. It is crucial for manufacturers to establish a biological evaluation plan in alignment with both ISO 10993 standard series and ISO 14971 requirements.
Manufacturers are also responsible for documenting their biological evaluation process. This documentation is essential to support the assessment of the medical device's biological properties, material selection, material characterization, and the verification of biological safety through a biocompatibility testing program laid out in the biological evaluation plan. The findings from the testing program should be compiled and discussed in a biological evaluation report.
It should be kept in mind that the biological evaluation process is not static; it must be revisited if there are changes in design or manufacturing processes, ensuring continued biocompatibility of the device. Additionally, post-market safety data might identify a potential risk and necessitate re-evaluation of biocompatibility. Manufacturers are also required to ensure that their biological evaluation, including their testing strategies, remains up-to-date with progressing standards or test methods.
Contact type and duration
One of the crucial steps in the beginning of biological evaluation process is identifying the intended anatomical location, frequency, and duration of exposure for the medical device. Devices that come into contact with internal body fluids or tissues for extended periods carry significantly higher biological risks compared to those that only touch intact skin for short durations.
One of the most critical points to consider when determining the contact duration of a medical device is its repeated or cumulative use. For devices expected or intended to be used multiple times, the categorization of the medical device should consider the potential cumulative effects, taking into account the duration and frequency of these exposures. This cumulative approach is particularly important for devices that frequently contact patient tissues, as repeated exposures can increase the risk of adverse biological effects.
After establishing the device's contact duration and type, the relevant biological endpoints can be selected according to Annex A of ISO 10993-1.
Material selection
The biological evaluation process also requires selecting the most appropriate materials during the design phase of medical devices. In the biological evaluation plan, each of these materials must be clearly documented and identified, including details such as full identification, composition, supplier information, and any colorants or additives used.
Even when the materials for the medical device are well-defined, there is no certainty that the final product is composed exclusively of these materials. The potential presence of substances introduced during the manufacturing process of the medical device should also be considered. Therefore, manufacturers must ensure their biological evaluation plan identifies both the medical device materials and the manufacturing processes.
Chemical characterization and toxicological risk assessment
ISO 10993-1 requires the identification of device components and manufacturing residues through chemical characterization in accordance with ISO 10993-18. This standard involves both qualitative and quantitative analysis of materials present in the finished device. The substances detected during this process are then assessed through a toxicological risk assessment, which takes into account factors such as the intended use, exposure conditions (e.g., invasiveness, duration), and the dose related to the medical device.
It is important to note that material characterization must be carried out before conducting any biological testing. The toxicological risk assessment, based on this characterization, can demonstrate the device's safety concerning various endpoints specified in ISO 10993-1 and may reduce the need for certain biological tests. These endpoints, which can be justified through toxicological risk assessment, include:
- Acute toxicity
- Subacute toxicity
- Subchronic toxicity
- Chronic toxicity
- Carcinogenicity
- Genotoxicity
- Reproductive and developmental toxicity
Chemical characterization and toxicological risk assessment offer several additional advantages. When a design change involving device materials (including a supplier change) occurs, the original biological testing data may no longer be applicable to the modified device or new materials. In such cases, it may be necessary to repeat the relevant tests outlined in ISO 10993-1. However, if chemical characterization data is available, the manufacturer can perform new characterization on the new device to scientifically validate equivalence, potentially avoiding the need to repeat biological tests.
Literature review
Another important aspect of the biological evaluation process is the literature review, which plays a critical role in both material selection and toxicological risk assessment. Conducting a literature review helps to determine whether the existing data in published studies is sufficient to establish the biological safety of the device, potentially eliminating the need for additional testing. Alternatively, it may reveal gaps in the available data, indicating that further testing is necessary.
Biological testing
Biocompatibility tests can be performed either in vitro or in vivo. In some cases, animal testing is necessary to provide evidence of biological safety. Wherever possible, efforts should be made to minimize animal testing by using alternative approaches, such as literature reviews, toxicological risk assessments that eliminate the need for animal tests, or opting for in vitro test methods.
In conclusion, very few medical devices are completely without risk. During the medical device design, the potential risks associated with their use must be weighed against the health benefits they offer to patients. Therefore, the results of any biological safety study must be interpreted within an appropriate benefit-risk context. The ISO 10993 standard series provide a structured framework for assessing the biological safety of medical devices, guided by risk management principles. This evaluation requires an understanding and expertise in chemistry, biology, as well as toxicology.


