

In this newsletter, we wanted to share basic information and terms about Clinical Research in medical devices that we guess you have started to hear frequently.
First of all, we would like to start by explaining why this burdensome body of work needs to be done:
Whether the potential risks of a medical device are at an acceptable level is determined by comparing them with the benefits it provides for its intended use. In order to measure this comparison accurately, clinical data on the product must first be collected to prove that the device offers adequate operating performance and is safe.
These collected data are then analyzed and submitted to the Notified Body in the form of a report as part of the Technical File, if it can be scientifically proven that the product provides more benefit than its risk if used in accordance with the declared intended use.
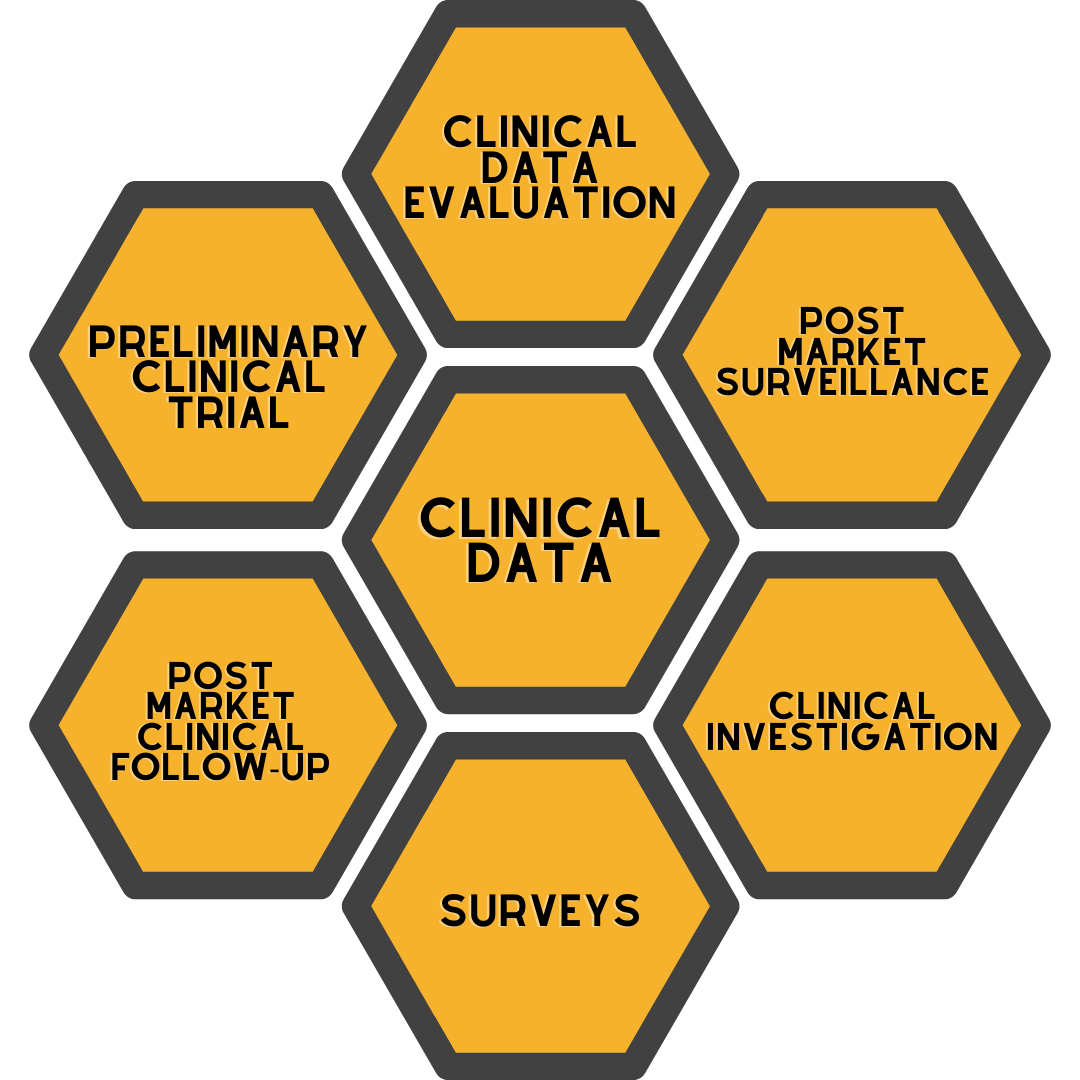
Clinical data, whose importance has increased with the MDR (2017/745) Medical Device Regulation, can be collected through the following items:
- Published clinical studies conducted with equivalent products of the medical device in question
- If the medical device in question is a product used in the market, feedback from patients, doctors and distributors of the product itself or adverse event reports / recalls of equivalent products
- Clinical studies conducted on the product before it is introduced to the market (Pre-Market) or after it is introduced to the market (Post-Market)
Until now, clinical evidence that the safety and performance of the Medical Device was adequate was mostly collected through literature research on “equivalent” products.
However, since the proof of "equivalent product" has become difficult after MDR (EU 2017/745), the need for evidence that the safety and performance of the medical device is sufficient to be generated through Clinical Research and used in the Clinical Data Evaluation report has increased.
Clinical Research
According to the standard "ISO 14155 - Clinical research of medical devices for humans", clinical studies are scientific studies carried out with volunteers and whose aim is to measure the safety and performance of the device in question on the specified intended use and target population.
Responsible of Clinical Research
The person, institution or organization responsible for initiating, conducting or financing the clinical trial is referred to as the Sponsor or Supporter. If a clinical trial is commissioned by the Sponsor, it is called a “Sponsored Clinical Trial”. Differently, clinical research initiated and carried out by university academics for their scientific research is classified as "Academic Clinical Research".
The application form and rules of the Ethics Committee and the Ministry for Academic and Sponsored Clinical Research are different. Even if the doctor conducting the research is an academician, if they are the founder of the company that will produce the product subject to research, the clinical research will be considered a Sponsored Clinical Research. In such cases, in order to maintain the impartiality of the research, auditing it by an independent committee may be a condition for obtaining approval.
Who manages the Clinical Trial?
Clinical Trial can be conducted by Sponsor's employees if they have the right team and experience. If there is no such team or experience; The sponsor may transfer all or some of its duties and authorities regarding the clinical trial to the "Contract Research Organization", briefly called "CRO", with a written contract. These institutions, whose English equivalent is CRO, can provide services at all stages from document preparation to study closure, or they can only carry out certain parts of the clinical research steps.
Design of Clinical Research
Clinical Research involves great responsibility as it is a study conducted on patients. In addition to the conscientious responsibility of risking patient health, the Sponsor also takes legal responsibility in accordance with the laws and regulations.
Apart from these, considering the use of serious human resources, examination and material use, reaching the number of volunteer patients and the follow-up period of the subject under investigation, it must be well designed as it requires a long period of time. Since incorrectly designed clinical studies will not provide sufficient and useful clinical evidence, the effort, time and financial investment spent on clinical research will be wasted. Therefore, your research should be carefully designed from the beginning.
Synopsis and Protocol of the Clinical Trial
There are different types of Clinical Trials. The type of Clinical Research is determined according to many parameters such as whether clinical data of the patients have been collected in the past, the status of the device (in the design phase or being sold), the purpose of use, the hypothesis to be proven by the study, and the targeted clinical data.
Clinical Research Protocol (Clinical Study Plan) includes all the details of the research to be conducted, the purpose of the study, how it will be conducted, which methods will be used, under what conditions and on what basis the volunteers will be included, ways to eliminate possible risks, if any, what the end points are, the follow-up period, the collected data. It is an important document that includes details such as how to analyze it statistically.
Synopsis can be considered as a summary of the Protocol and does not contain much detail other than providing general information.
Since the purpose of clinical research, the criteria to be measured, the medical methods to be used, the place of use and indication of the device, and the patient profile will differ for each clinical research, a separate protocol is prepared for each clinical research and separate approval is obtained from the authorized institutions for each research.
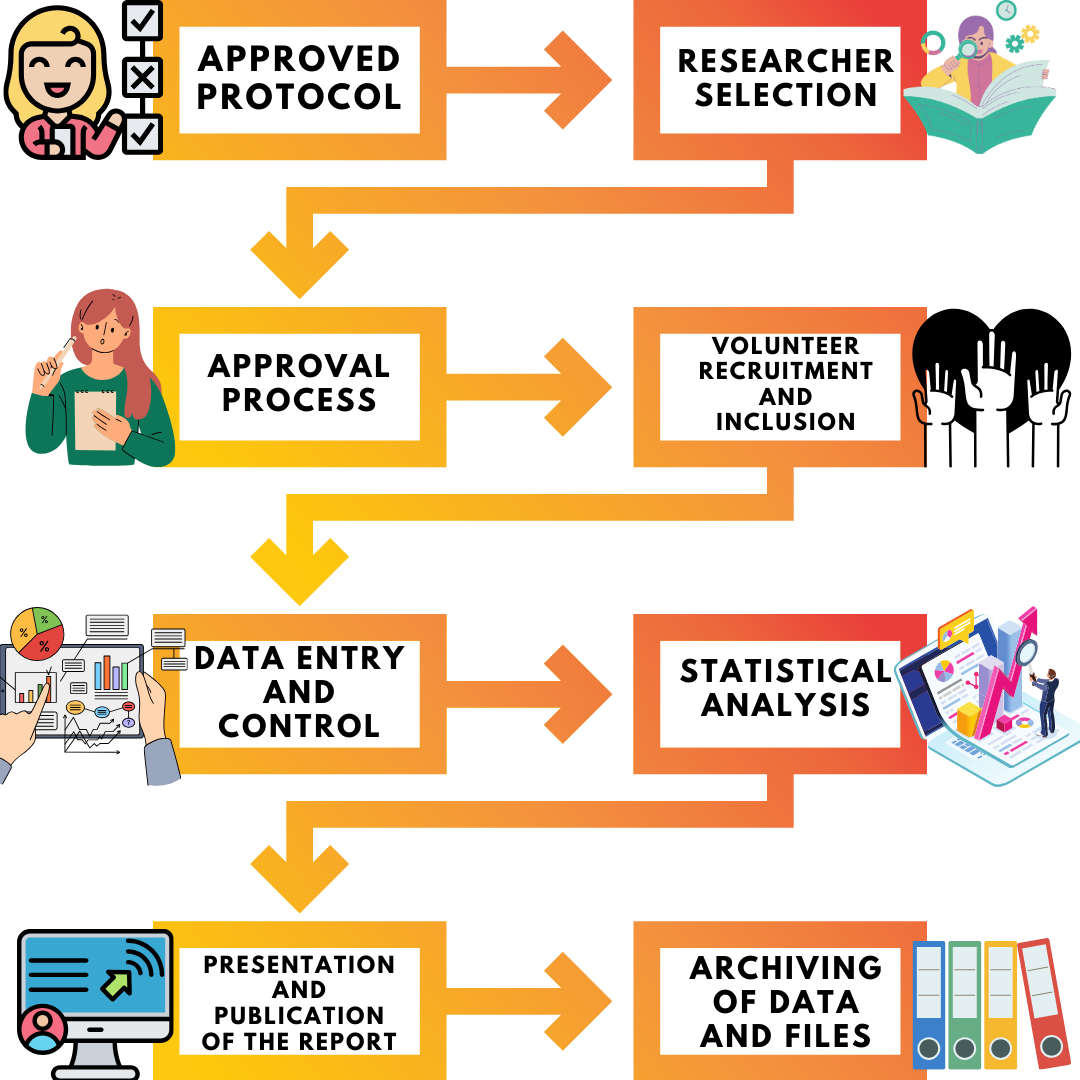
Clinical Research Steps
The basic steps of a clinical trial consisting of many items are as follows;
- Preparation of Clinical Research Protocol
- Preparation of Clinical Research documents (Case Report Form, Informed Volunteer Consent Form, Volunteer Information Form, Researcher Brochure, Ethics Committee Application Form, Ministry Application Form, Adverse Event Information Form, etc.)
- Selection and agreement of the centers where the clinical research will be conducted and the doctors who will conduct the research.
- Applying to competent institutions and authorities (Ethics committee and Ministry of Health) and obtaining their approval,
- After receiving research approval, the team that will take part in the study is formed in each center and the study is initiated by providing the necessary information and training.
- Initiating the volunteer recruitment of the clinical trial and completing the volunteer recruitment by reaching the number of volunteers determined in the protocol.
- Conducting the clinical trial within the framework of the protocol
- Monitoring and auditing of clinical research
- Terminating and closing the clinical trial
- Preparation of the final report by analyzing the data obtained from the clinical research according to statistical methods
- Preparation of the content of clinical research results for congress presentation and publication in scientific (peer-reviewed) journals
- Archiving of data by filing
Selection of the center where the research will be conducted is another important issue in medical device studies. Choosing centers where the targeted volunteer population is dense and where there are researchers experienced in the treatment in which the device is used will ensure that the research is carried out more easily and the targeted number of volunteers is reached in a short time.
Another important issue is the physician or dentist called the Chief Investigator or Responsible, who have completed their training in the specialty related to the research topic and is responsible for the conduct of the research.
Experts who are especially experienced in the treatment method in which your device is used and in the use of your device will make a positive difference in successfully measuring product performance. It should not be forgotten that the researcher and the clinical research team must have received "Good Clinical Practices" (GCP) training, and if they are missing, they must be completed.
In order to facilitate the selection of centers and researchers in your clinical research, DeSia provides support by making preliminary agreements with hospitals.
Change in Clinical Trial
No changes can be made to the principles stated in the clinical research protocol after obtaining the approval of the ethics committee and subsequently the ministry. In order for any changes to be made to the clinical study protocol, the amended protocol must be submitted to the approval authorities and re-approval must be obtained. For this reason, the design phase and protocol preparation must be carried out meticulously.
Patient relations during the Clinical Research process
The selection of patient volunteers is carried out according to the criteria specified in the protocol, and each patient who agrees to volunteer is required to sign the "Informed Volunteer Consent Form", which is extremely important in legal terms. While volunteers can only be included in the study after signing this form, it should be noted that they have the right to withdraw their consent and withdraw from the study at any time.
Informed Voluntary Consent Forms should contain specific information prepared specifically for the research, therefore consents obtained upon admission to the hospital that contain only general statements such as "I approve the use of patient data and examinations in clinical research" should not be used.
When findings regarding negative or undesirable effects on Volunteer Patients are encountered during the research process, these adverse events (Adverse Event - AE) are collected and an "Adverse Event Report" is prepared, and the Ethics Committee and the Competent Authority are notified at certain intervals depending on their level of seriousness. If necessary, the clinical trial is terminated.
Clinical Research Cost
Clinical studies are studies that require high-caliber employees such as doctors, nurses, statisticians, materials, tools and facilities used in medical operations and examinations, and may have high expenses due to the costs of different institutions such as CRO, approval bodies and insurance companies.In some cases (pre-market studies, Interventional Research, etc.), it may be required that patients be covered by private health insurance. In such a requirement, we provide services from our existing business partners to insure the research.

Once the Clinical Trial is completed
The collected data is compiled and analyzed within the framework of statistical models. Based on this analysis, the Clinical Research Final report is prepared.
Researchers, due to their academic careers, and Sponsors, due to both CE certification requirements and evidence-based data becoming increasingly important, generally request articles and presentations for congresses about the research topic and results in order to increase the awareness of their products by being included in congress and medical literature. As in all steps, DeSia also provides services in these preparations.
Clinical Research can be grouped into two main headings in terms of design:
Retrospective Studies: Based on retrospective data collection. In order for this study to be conducted, the data of patients who have previously been treated with the device in question must be available in the centers in accordance with the intended study.
Prospective Studies: Patient data are collected prospectively. Volunteer patients are included in the study and receive treatment with the device in question after all approvals are obtained.
It is generally possible to categorize Prospective Studies into two main groups: "Observational" and "Interventional".
a. Observational: In observational clinical studies, researchers follow volunteers within a research plan/protocol.
Volunteers can receive interventional treatment involving medical devices within the framework of routine treatment, but researchers cannot use a special interventional treatment outside of the routine. The study must be performed based on the approved indication of the product.
The product must be used routinely.
b. Interventional: In interventional clinical studies, researchers follow volunteers within a research plan/protocol.
While volunteers receive an interventional treatment in which a medical device will be used, the researcher may perform tests and operations outside the routine to answer specific questions targeted in the study (e.g. special patient population: children, elderly, diabetic, etc.).
An existing device can be compared with a new device, two different treatment methods can be compared, specific questions can be answered with different interventions/imaging.
In conclusion, the intricate world of Clinical Research in medical devices is not only a regulatory necessity but also a testament to the commitment of researchers, manufacturers, and healthcare professionals to ensure the highest standards of safety, efficacy, and innovation for patients worldwide. As the landscape continues to evolve, particularly with the introduction of regulations like the MDR (2017/745), understanding the intricacies of clinical trials becomes paramount. At DeSia, we remain dedicated to supporting and guiding our partners through every phase of clinical research, ensuring that the journey from conception to market is paved with knowledge, expertise, and a steadfast dedication to excellence. Together, we can shape a brighter, safer future for healthcare and the countless lives it touches.


