

![]() Avrupa Birliği'ne tıbbi cihaz sağlayacak firmaların ve ilgili tüm tarafların gerekli hazırlıkları yapması için Mayıs 2017’de yayınlanan ve Mayıs 2020’de yürürlüğe girecek olan Tıbbi Cihaz Tüzüğü, (MDR – Avrupa Birliği Tıbbi Cihaz Regülasyonu 2017/745/EEC) tıbbi cihaz üreticilerine birçok ek sorumluluk getirmiştir. Bu sorumluluklar arasında hem zaman alıcı hem de maddi yükümlülük oluşturan sorumluluklardan birisi de ürüne ait “Klinik Veri” sağlamaktır.
Avrupa Birliği'ne tıbbi cihaz sağlayacak firmaların ve ilgili tüm tarafların gerekli hazırlıkları yapması için Mayıs 2017’de yayınlanan ve Mayıs 2020’de yürürlüğe girecek olan Tıbbi Cihaz Tüzüğü, (MDR – Avrupa Birliği Tıbbi Cihaz Regülasyonu 2017/745/EEC) tıbbi cihaz üreticilerine birçok ek sorumluluk getirmiştir. Bu sorumluluklar arasında hem zaman alıcı hem de maddi yükümlülük oluşturan sorumluluklardan birisi de ürüne ait “Klinik Veri” sağlamaktır.
Bu zamana kadar birçok tıbbi cihazın onay aşamasında Klinik Veri Değerlendirmesi yapılırken ürünün güvenlik ve performansını kanıtlamada eşdeğer ürünlere ait klinik veriler kullanılmıştır. Yeni yasa, tıbbi cihaz üreticilerine bu konuda MDR’ın 61. maddesi ile ciddi kısıtlamalar getirerek; üreticilerin kendi ürünlerine ait klinik veri oluşturmalarını istemektedir.
“Klinik Veri”nin toplanması uzun bir zaman aldığı için bu konudaki eksikliklerinizi belirleyip doğru çalışmaları hemen başlatmak, sonraki çalışmalarınızda zamanlama ve maliyet açısından önemli avantajlar sağlayacaktır.
Yeni tüzükle (MDR) birlikte, eski direktiften (9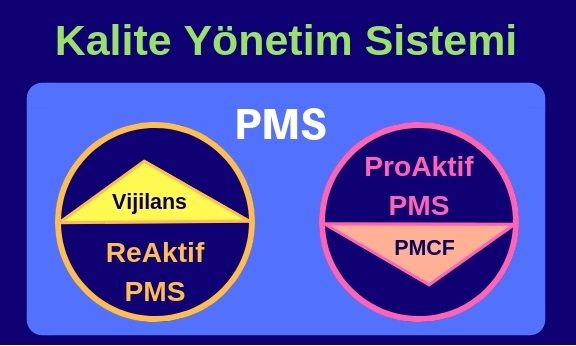 3/42/EEC MDD, 90/385/EEC AIMDD) farklı olarak klinik veri oluşturulması ve/veya toplanması ürünün onay aşamasında planlanıp tamamlanan bir süreç olmaktan çıkmıştır. Klinik verinin sağlanması, ürünün yaşam döngüsü boyunca Pazar Sonrası İzleme (PMS) ve Pazar Sonrası Klinik Takip (PMCF) aktiviteleri ile periyodik olarak tekrar edecek bir çalışma olmalıdır. PMCF (Pazar Sonrası Klinik Takip) planı ve aktiviteleri MDR’ın birçok farklı bölümünde istenmekte ve bu çalışmaların sonuçlarıyla Klinik Veri Değerlendirmenin (KVD) beslenmesi gerektiğinin özellikle altı çizilmektedir.
3/42/EEC MDD, 90/385/EEC AIMDD) farklı olarak klinik veri oluşturulması ve/veya toplanması ürünün onay aşamasında planlanıp tamamlanan bir süreç olmaktan çıkmıştır. Klinik verinin sağlanması, ürünün yaşam döngüsü boyunca Pazar Sonrası İzleme (PMS) ve Pazar Sonrası Klinik Takip (PMCF) aktiviteleri ile periyodik olarak tekrar edecek bir çalışma olmalıdır. PMCF (Pazar Sonrası Klinik Takip) planı ve aktiviteleri MDR’ın birçok farklı bölümünde istenmekte ve bu çalışmaların sonuçlarıyla Klinik Veri Değerlendirmenin (KVD) beslenmesi gerektiğinin özellikle altı çizilmektedir.
Tıbbi Cihaz Tüzüğü (MDR), Ek XIV Kısım B’ye göre tıbbi cihaz üreticilerinden PMCF planı hazırlarken aşağıdaki konuları karşılamalarını beklemektedir;
– PMCF sırasında izlenecek olan genel metot ve prosedürleri içermesi. (Klinik veri ne şekilde üretilecek?)
– PMCF sırasında izlenecek olan spesifik metotları içermesi. (PMCF çalışmaları, retrospektif ve/veya prospektif çalışma ön detayları, anket çalışmaları vb…)
– İzlenecek metodun uygunluğunun tartışılması.
– Klinik veri değerlendirme raporuna ve risk değerlendirmesine atıf.
– Eşdeğer (MDR çerçevesinde eşdeğerlik kanıtlanabiliyor ise) ve/veya benzer cihazların klinik verilerinin değerlendirilmesi.
– Ürüne özel spesifikasyon ve standart var ise belirtilmesi.
– PMCF aktiviteleri için detaylandırılmış zaman çizelgesi.
Klinik veri toplama aktivitelerinde önemli bir payı olan PMCF çalışması için;
- PMCF planının içeriği
- Takip için yapılması gereken klinik çalışmalar,
- Hangi verilerin eksik olduğu ve toplanması gerektiği
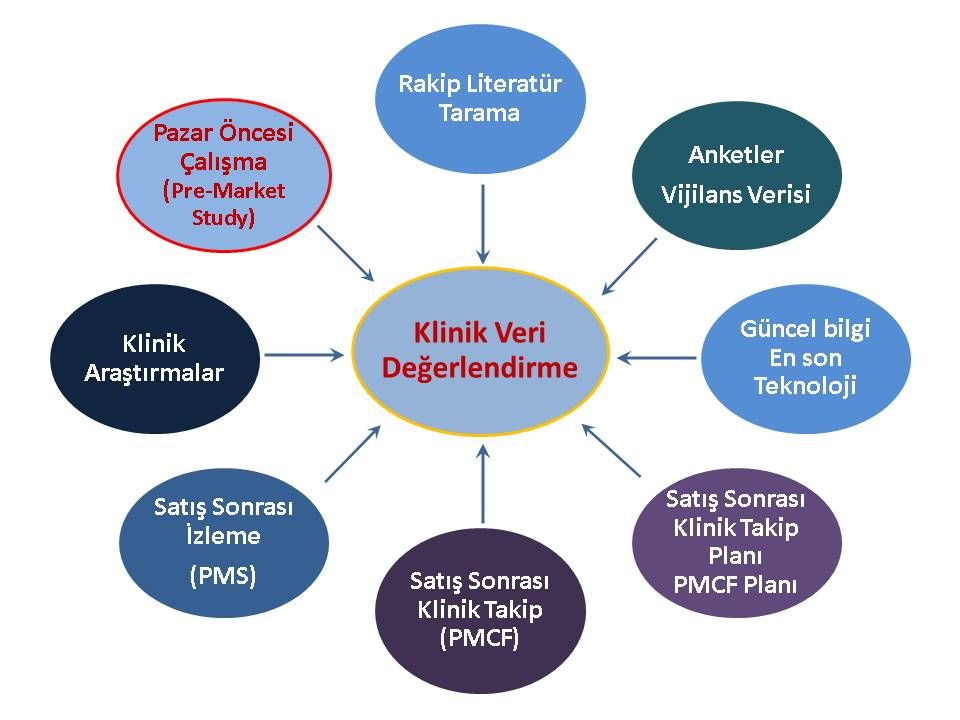
“Klinik Veri Değerlendirme (KVD)” raporunda netlik kazanacaktır. (Veri eksikliği olan özel bir endikasyona yönelik veri tamamlama mı yapılacak? veya mevcut güvenlik ve performans kanıtlama sadece eşdeğerlik üzerine yapıldıysa ürüne ait klinik veri oluşturmaya gerek var mı? vb…) Bu nedenle ilk adım olarak Klinik Veri Değerlendirmenize geri dönerek MEDDEV 2.7.1 Rev 4’e tamamıyle uyduğuna emin olmanız ve Tıbbi Cihaz Tüzüğü’nü (MDR) göz önüne alarak gerekirse sonuçlarının yeniden değerlendirilmesi çok önemlidir.
KVD raporunuzun sonucunda belirleyeceğiniz veri açıklarına istinaden bir PCMF planı hazırlayarak; bu plan çerçevesinde bir an önce proaktif bir şekilde veri toplamaya başlamanız yararlı olacaktır.
Mevcut klinik verinizin değerlendirmesi sonucunda, bir klinik çalışma yürütülmesi gerekli ise yasa ve standartların gerekliliklerine uygun olarak çalışma protokolünün (klinik araştırma planı) yazılması, araştırmacıların / merkezlerin seçilmesi ve Etik Kurul ve Sağlık Bakanlığı onaylarının alınması aşamalarının titizlikle takip edilmesi gerekmektedir.
Klinik araştırmalarda en önemli faktörlerden biri de araştırmanın hem ürüne hem de amaca yönelik doğru tasarlanmasıdır. Doğru tasarlanmayan bir çalışma, hem zaman ve kaynak kaybına neden olacak, hem de ürün için güvenlik ve performansı kanıtlayıcı bilgiyi topla(ya)mayacaktır.
Tıbbi Cihaz Tüzüğü’nün yürürlüğe girmesi için yaklaşık 1 yıllık bir süremiz kaldı. Bu nedenle en kısa zamanda elinizdeki klinik verinin durumunu analiz ederek, yukarıda verdiğimiz bilgilere göre gereken doğru çalışmaları başlatmak, MDR hazırlıklarının en önemli bölümünü oluşturmaktadır.
Ürünlerinizin riski ve mevcut durumu göz önüne alınarak klinik verinizi oluşturmak adına size en uygun klinik çalışma tasarımının seçilip uygulanmasında AdviQual ve klinik araştırmalar şirketimiz DeSia her zaman destek vermeye hazırdır.


